Alport Syndrome
Alport Syndrome is a genetic rare disease occurring from mutations in the genes that code for type IV collagen. The type IV collagen alpha chains are primarily located in the kidneys, eyes, and cochlea (the inner ear), thus the condition is characterized by kidney disease, loss of hearing, and eye abnormalities. Eventually, patients present with proteinuria (elevated protein in the urine), hypertension, progressive loss of kidney function (gradual decline in GFR), and end-stage renal disease (ESRD).
Approximately 67,000 individuals in the United States are estimated to be affected by this disorder, which is a notable contributor to chronic kidney disease (CKD). Among adolescents and young adults, it frequently progresses to end-stage renal disease (ESRD). This condition represents 1.5% to 3.0% of pediatric cases requiring renal replacement therapies in both the European Union and the United States.
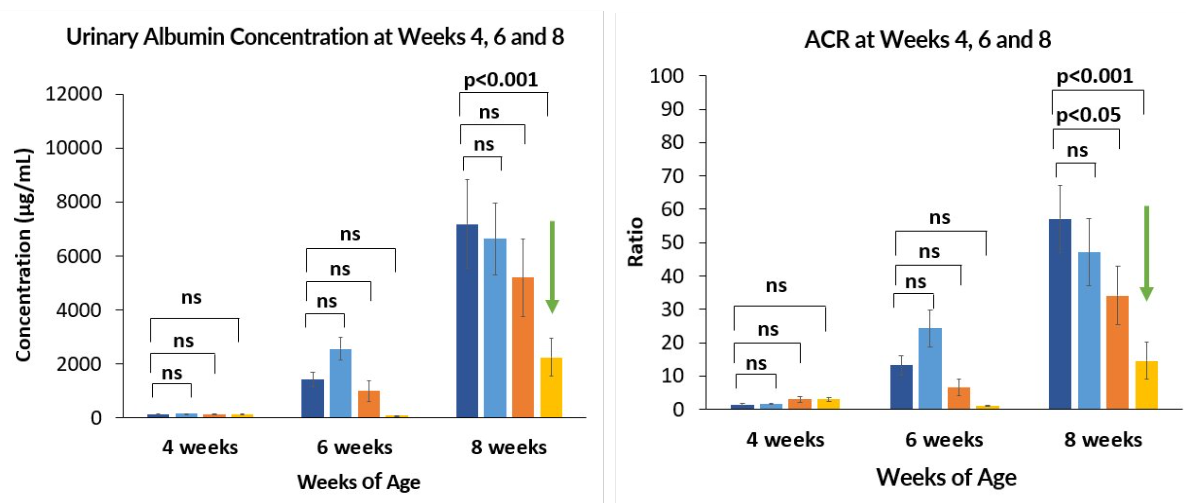
Setanaxib and Ramipril combination significantly reduced fibrosis and the rate of decline in glomerular function
The Col4a3 KO mouse model (n=40) was used to assess the potential of setanaxib alone and in combination with ramipril (SoC).
Setanaxib + ramipril combination vs. placebo 8 weeks:
- Highly statistically significant reduction in albuminuria and urine ACR
- Statistically significant reduction in glomerular sclerosis
- In silico analyses showed increased glomerular basement membrane and collagen proteins
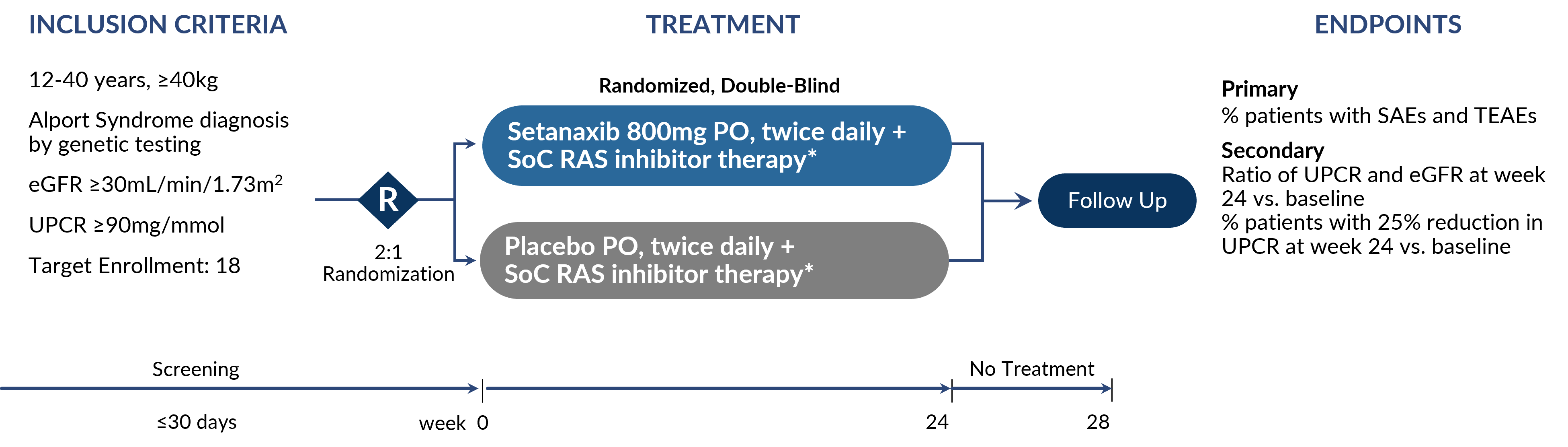
Phase 2 RCT for Alport Syndrome
There are no approved products for Alport Syndrome. The US FDA and EMA granted orphan drug designation (ODD) for the treatment of Alport syndrome with setanaxib in September and October of 2023, respectively. Ph2 randomized, controlled trial was initiated in November 2023.